
Review article: Regulation of neuronal cell death and survival
- Richard Norris
- Sep 18, 2016
- 16 min read
To start off my blog I have decided to post a review article that I wrote a couple of years ago. This post maybe of most interest to those who work in the field or who are familiar with the concepts described. For subsequent posts I shall endeavor to write more for the general reader.
Regulation of neuronal cell death and survival (with focus on survival signal withdrawal induced apoptosis in sympathetic neurons)
The regulation of neuronal apoptosis is crucial to the establishment of a correctly wired nervous system during development. It has long been known that specific populations of neurons depend on a limited supply of neurotrophins for survival, but the mechanisms controlling this process remain incompletely understood. In this review I discuss some of the key components of trophic signal-dependent survival of neurons in the peripheral nervous system, the apoptotic pathways resulting from the loss of signal, and the regulatory mechanisms controlling them. I also highlight some areas where there is uncertainty over the precise mechanisms involved, the alternative models proposed and the evidence supporting them.
Introduction
Neuronal apoptosis during embryonic and post-natal development is an important mechanism which ensures neuronal populations of the correct size are established, and that neurons forming inappropriate connections to the targets they are innervating are removed (Kristiensen et al 2011). Apoptosis also occurs in neurons following injury to the nervous system or in neurodegenerative disorders, therefore an understanding of the molecular pathways underlying this process may prove important to the development of therapies.
During the development of the peripheral nervous system in mammals approximately half of the neurons formed die by apoptosis. Survival of these neurons depends on their acquisition of a limited supply of trophic factors. The neurotrophic theory states that the fate of developing neurons is regulated by the availability of target-derived neurotrophic factors (Levi-Montalcini, 1987). These neurotrophins act as survival signals to counteract the intrinsic apoptotic tendency of neurons. It has long been known that neurons in the peripheral nervous system require such survival signals to prevent their death during development. However, despite the evolutionary conservation of significant aspects of the neuronal apoptosis signalling, particularly in terms of the involvement of caspases, the absence of a trk gene (encoding one of the key receptors involved in regulating neuronal survival) in either C. elegans or D. melanogaster has proved a major stumbling block in the quest to understand neurotrophin receptor signalling (Nikoletopoulou et al, 2010). Another limiting factor in studying neuronal cell death is the lack of availability of neuronal cells for in vitro experiments. In recent years this problem has been overcome to some extent by technological advances, including microarray technology, significantly improving the potential output of individual assays.
In the process of elucidating the precise ligand/receptor interactions and intracellular pathways that lead to cell death or survival an important question to ask is why only certain populations of neurons are dependent on NGF for survival. Perhaps the most established model of neurotrophin-induced survival signalling involves a signal initiated in distal axons transmitted in a retrograde manner and activated by NGF binding to the TrkA receptor, resulting in pro-survival receptor tyrosine kinase mediated signalling (Harrington et al, 2011)
In neurons, as well as many other mammalian cell types, the key event in apoptosis is the release of cytochrome c from the mitochondrial inter-membrane space into the cytosol. Once released, cytochrome c initiates the ATP-dependent autocatalytic processing of procaspase 9 via its interaction with apoptotic protease activating factor 1 (Apaf-1). Caspase 9 then activates the effector caspases, including caspase 3 (Li et al 1997). However, in sympathetic neurons exposed to Nerve growth factor (NGF), cytochrome c alone is not sufficient to induce apoptosis. These neurons must become ‘competent to die’ a process that only occurs following NGF-deprivation (Deshmukh and Johnson, 1998).
Pro/mature neurotrophins
The neurotrophic ligands NGF, Brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT3), are synthesized from precursors (proNGF, proBDNF and proNT3). These proneurotrophins also activate signalling pathways in target cells, but often the downstream effects are distinctly different (Song et al 2010). ProNGF is converted into the active neuroprotective NGF via post-translational cleavage of the pro-domain, but while the anti-apoptotic function of mature NGF is well established, the pro-domain may induce apoptosis via p75NTR and the JNK/MEK1/2 pathway (Armugam et al 2012), though the evidence is divided on this point. While some studies support the claim that mature NGF transmits cell survival and death by binding to TrKA/ p75NTR, while proNGF induces apoptosis via p75NTR (Chao et al 2003; Lee et al 2001; Harrington et al 2004), more recent studies have demonstrated that proNGF may promote cell survival, albeit to a lesser extent than mature NGF (Clewes et al, 2008; Sobottka et al, 2008). It has also been demonstrated that the function of proNGF is determined by the relative expression levels of the TrkA and p75NTR receptors. A lower ratio of trkA/ p75NTR expression results in pro-apoptotic signalling, whilst high ratios lead to pro-survival signalling (Masoudi et al 2009). This last explanation sounds like the most logical explanation for the alternative functions of the NGF pro-domain and could account for the discrepancy in findings from the various studies as these ratios may not have been taken into account. proNGF has also been found to initiate a pro-apoptotic signal via interaction with the sortillin receptor (Jansen et al.,2007; Nykjaer et al., 2004; Song et al., 2010).
The pro-apoptotic signalling initiated by proNGF acts via the JNK/MEK1/2 pathway and may also be mediated by Bim and the Bcl-2 protein family, indicating the involvement of similar factors to mature NGF signalling (Armugam et al 2012). ProNGF also blocks survival signalling in basal forebrain neurons via activation of Akt and ERK indicating it plays a significant role in neural development. In CNS neurons PTEN (phosphatase and tensin homolog deleted on chromosome 10) acts as a switch regulating the balance between proneurotrophin/p75NTR induced apoptotic signalling and mature neurotrophin/Trk induced survival signalling (Song et al 2010).
Receptors
As eluded to above, cellular responses to signalling by both mature and precursor neurotrophins occur via three distinct classes of receptor, the tyrosine kinase receptor tropomyosin receptor kinase A (TrkA), p75NTR and sortilin (Armugam et al 2012). The combination of receptors and pro/mature neurotrophic ligands dictate and regulate the neuron by promoting its survival or by inducing an apoptotic pathway resulting in cell death (Nykjaer et al, 2004; Sobottka et al, 2008; Song et al, 2010).
The importance of TrkA was established using knock-out studies of sympathetic neurons isolated from the murine superior cervical ganglion at E17, a critical stage of neuronal development during which multiple neuronal populations become dependent on NGF for survival. In the absence of NGF, wild-type neurons were rapidly eliminated, whereas the majority of TrkA-/- neurons survived. Curiously though, the addition of NGF to TrkA-/- neurons resulted in rapid cell death, suggesting the involvement of other receptors in NGF-mediated signalling (Nikoletopoulou et al., 2010). These observations also challenge the assertion that neurotrophins only act positively via kinase-dependent signalling.
NGF/TrkA survival signalling is largely limited to sympathetic neurons and such apoptotic pathways are not found in the CNS with the exception of the cholinergic neurons, a small population neurons in the basal forebrain (Sobreviela et al., 1994), a proportion of which require NGF for survival (Hartikka & Hefti, 1988; Müller et al., 2012)). A possible explanation for this is that the expansion of the Trk family of receptors occurred during the evolutionary segregation of the peripheral and central nervous systems. TrkA and TrkC mediate survival/apoptotic signals whereas TrkB (expressed in the CNS) does not. (Nikoletopoulou, 2010). NGF and NT3 activate TrKA in a similar manner but only NGF initiates retrograde survival signalling via TrkA in sympathetic neurons. Upon neurotrophin binding TrkA is activated by auto-phosphorylation and internalized. NT3/TrkA complexes are also internalized but do not form endosomes able to be transported by the actin cytoskeleton and thus fail to mediate signal transduction (Harrington et al 2011)
One question mark concerning the standard neurotrophic theory is based on examination of the ‘default apoptotic state of developing neurons. If the neurotrophin receptors are acting as dependence receptors (capable of inducing cell death in the absence of their ligand (Bredesen, Mehlen and Rabizadeh, 2005) then inactivation of the ligand should result in a more severe phenotype than inactivation of the receptor. But is the loss of survival signals sufficient to account for neuronal death, or is an active pro-apoptotic activity on the part of the receptor necessary to create the ‘‘intrinsic apoptotic status’’ in the absence of neurotrophic survival signalling? (Tauszig-Delamasure et al 2007). Support for this concept comes from the observation that ‘TrkA causes cell death of neurons by virtue of its mere expression, and this death-inducing activity is prevented by addition of NGF’ (Nikoletopoulou et al., 2010).
De novo gene expression and regulation
NGF withdrawal activates the MLK-JNK-c-Jun pathway and inactivates the PI3K-Akt and Raf-MEK-ERK pathways in a complex signalling cascade. Inhibitors of RNA and protein synthesis (actinomycin D and cyclohexamide) block NGF-deprived apoptosis indicating that de novo gene expression is an important factor in survival signaling (Lipscomb et al 1999). This led to an important prediction, in that the expression of specific genes is altered following NGF withdrawal, and that the ‘proteins encoded by these genes trigger cell death.’ (Kristiensen et al, 2011). A number of key genes were identified by expression studies of candidate genes (c-jun, bim, tp63, puma) or by using mRNA differential display (egln3). It was discovered that mRNA and protein levels of these genes increases after NGF withdrawal and these results were confirmed by observing the knock-out phenotype in mice. However, only a small number of genes responsible for cell death are differentially expressed following NGF-withdrawal, and in sympathetic neurons the components of the intrinsic apoptotic pathway (bax, cytochrome c, apaf-1 and caspase-9) are not significantly affected. The most significant genes in terms of expression levels are the BH3-only proteins responsible for activating the intrinsic (mitochondrial) apoptotic pathway: Bim, Puma, dp5 and Bmf (Kristiensen et al 2011).
Bim:
The Bcl-2 family protein Bim interacts with both the anti-apoptotic Bcl-2 proteins and the multidomain pro-apoptotic effector proteins Bax and Bak and is therefore important in initiating and regulating the intrinsic pathway. (Hughes & Ham, 2011). In sympathetic neurons exposed to NGF, overexpression of Bim results in the release of cytochrome c and the induction of apoptosis, indicating that the strong pro-apoptotic signalling of Bim is able to override survival signalling. Neurons in Bim-/- knockout mice are largely protected from neurotrophin withdrawal induced death (Whitfield et al, 2001; Putcha et al, 2004; Coultas et al, 2007).
The regulation of bim expression in neurons is complex and remains incompletely understood. It involves a number of different mechanisms and several transcription factors. This level of regulation is important as inappropriate expression of bim may result in the unwanted loss of neurons and a deleterious phenotype. Three transcriptional pathways targeting elements of the bim promoter have been described (Biswas et al 2007), as well as post-translational modifications. Bim transcription is down-regulated by PI3-K-Akt signalling via FOXO binding sites, and up-regulated by the Cdk4-E2F-Myb pathway via Myb binding sites in the bim promoter. Perhaps the most important mechanism of Bim up-regulation in sympathetic neurons involves MLK-JNK-c-Jun signalling. Overexpression of dominant negative c-Jun protein reduces the increase in bim expression resulting from NGF withdrawal (Whitfield et al, 2001; Hughes & Ham, 2011).
So if bim is a common target for three different neurotrophin deprivation-induced transcriptional pathways then the question of whether this regulation occurs independently and therefore in a redundant system or in a cooperative or additive mechanism is raised. Promoter-reporter assays in which FoxOs, c-Jun and Mybs were sequentially knocked down support a model in which bim induction following NGF withdrawal is dependent on simultaneous activation of all three transcription factors. As such, the bim promoter is acting as a coincidence detector which does not induce bim expression unless all three regulatory sites are occupied simultaneously (Biswas et al 2007). This conclusion is supported by the more recent discovery of the role of the hetero-trimeric transcription factor NF-Y. NF-Y bound to the conserved inverted CCAAT box (ICB) in the bim core promoter in the presence of NGF recruits transcriptional co-activators CBP and p300. Again dominant negative mouse mutants were used to establish the requirement of NF-Y activity for bim promoter function. Upon NGF withdrawal FOXO3a also recruits CBP/p300 (to two conserved sites flanking the ICB) and a stable multi-protein complex is formed, possibly including c-Jun and Myb, which increases bim expression. These findings provide a model of protein-protein interactions at the bim promoter in which different signalling pathways act cooperatively to control the expression of bim following neurotrophin withdrawal (Hughes & Ham, 2011) (figure 1).
Another proposed mechanism of regulation of Bim-dependent apoptosis is the multi-site phosphorylation of the Bim protein (Ley et al 2005). Knock in mice were created with germ-line mutations in which key phosphorylation sites were substituted with Ala residues. Differential phosphorylation of Bim affected protein stability as well as the interaction of Bim with other Bcl2 family proteins. These studies established Bim phosphorylation as a control mechanism able to set the sensitivity and threshold response of Bim activation in response to pro-apoptotic stimuli (Hübner 2008). One may conclude from this evidence that this level of Bim regulation acts as a fail-safe mechanism ensuring that neuronal cells are only directed down an apoptotic path when a series of conditions have been met. The promoter regulation and post-translational phosphorylation mechanisms appear to act as an ‘on/off’ switch and a fine tuning mechanism respectively.

Figure 1. Model of transcriptional regulation of bim expression involving survival signalling in sympathetic neurons (Hughes et al, 2011)
Puma and dp5
The dominant negative mutants generated to study the effect of NF-Y on the bim promoter afforded only partial protection (~40% of cells were viable 72 hours after the withdrawal of NGF). The likely explanation for this is that Puma and Dp5, can partially compensate for the loss of Bim (reviewed in Putcha and Johnson 2004). Like bim, it appears that control of p53 up-regulated modulator of apoptosis (Puma) induction is regulated by multiple signalling pathways. Studies of puma have focused on its role in trophic factor deprivation in cerebellar granule neurons (CGNs) (Ambacher 2012) and in apoptosis induced by DNA damage in sympathetic neurons (Wyttenbach and Tolkovsky 2006), however the findings may point to key mechanisms involved in survival signal withdrawal in the developing peripheral nervous system.
In CGNs the expression of puma was reduced significantly by Glycogen synthase kinase 3β (GSK3β) inhibitors which in turn reduced the level of trophic factor deprivation-induced apoptosis. GSK3β has been shown to be inhibited by AKT-mediated phosphorylation of serine-9 (Dudek et al 1997) indicating that PI3K-AKT inactivation induces puma expression and neuronal apoptosis through a GSK3β -dependent mechanism (Ambacher 2012). Using a similar approach it was also found that inhibition of JNK significantly reduced Puma expression and apoptosis. So do these signalling pathways converge in the regulation of puma induction, as in the regulation of bim, or do they act independently? Here the picture is less clear. Western blot analysis showed that inhibition of GSK3 had no effect on the trophic factor-induced up-regulation of the JNK pathway, and that phosphorylation of GSK3β and AKT signalling was unaffected by JNK inhibition, suggesting that these pathways act independently in the regulation of puma. However, FoxO3a has also been implicated in puma induction in growth factor withdrawal-induced apoptosis (You et al 2006), and examination of the requirement of FoxO3a in this role in CGNs does suggest some convergence. The requirement of FoxO3a for puma induction was determined by FoxO3a knockdown experiments which showed a significant decrease in puma expression. Further analysis showed that JNK and GSK3β can regulate FoxO3a via an indirect mechanism or via an AKT-independent mechanism (Ambacher 2012). So clearly there is evidence of cooperation between signalling pathways in the regulation of puma, although further studies are required to determine the precise mechanisms.
Dp5 expression is also regulated by the MLK-JNK pathway in response to NGF withdrawal. The sequences regulated in the dp5 gene were identified using reporter constructs with each region of dp5 linked to luciferase. 1kb of promoter sequence, a short sequence from the 18kb intron and the entire 3’UTR were activated, however the construct containing all three regions induced the highest dp5 expression level indicating, as seen in the other BH-3 only genes, combinatorial regulation. Microinjection of MLK inhibiting proteins reversed this induction of dp5 confirming results from previous studies (Harris & Johnson 2001). The same result was observed using JNK inhibitors suggesting that transcriptional regulation occurs via JNKs. Dp5 also contains a conserved ATF-binding site which is able to bind c-Jun and ATF2. Site-directed mutagenesis to abolish the dp5 AP-1 binding significantly reduced NGF withdrawal-induced promoter activity. Finally, overexpression of the c-Jun dominant negative mutant JunΔ169 reduced dp5 expression suggesting that dp5 is regulated by AP-1 and activated by the MLK-JNK pathway via its ATF site (Towers et al 2009).
Although there is significant variation in the mechanisms controlling each of the key BH3-only pro-apoptotic proteins, and in some cases much still to be elucidated, what is clear is that developing neurons employ multiple mechanisms to regulate their expression and there is strong evidence of cooperation between pathways targeting key regulatory sites in each gene. The MLK-JNK pathway appears to be of critical importance, acting via c-Jun to induce expression of bim, puma and dp5 following NGF withdrawal (figure 2).
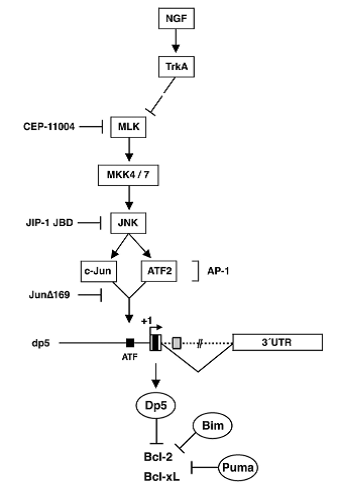
Figure 2. Regulation of dp5 by the MLK/JNK pathway, mediated by c-Jun and ATF2 (Towers et al, 2009)
Conclusions/future work
As I have described the neurotrophin-dependent model of selective sympathetic neuronal survival has been established following decades of research, and in recent years much of the complex signalling involved in regulating apoptosis in developing neurons has been elucidated. From the neurotrophin/receptor interactions to the regulation of the induction of proapoptotic BH3-only pro-apoptotic proteins by multiple signalling pathways. But much is still left be understood and future studies will hope to yield further insights into this important area of development. New technologies have made possible the rapid discovery of targets involved in survival/cell death signalling and work is now required (perhaps candidate gene studies) to investigate these further. Activation of the c-Jun pathway has been linked to Alzheimer’s and Parkinson’s (Okazawa & Estus, 2002 and Peng & Anderson, 2003) and therefore perhaps further discoveries in this field will also contribute to our understanding of the molecular mechanisms of neurological disease.
References
Ambacher, K.K., Pitzul, K.B., Karajgikar, M., Hamilton, A., Ferguson, S.S., Cregan, S.P., 2012.
The JNK- and AKT/GSK3β- signaling pathways converge to regulate Puma induction and neuronal apoptosis induced by trophic factor deprivation. PLoS One, 7(10) e46885. doi: 10.1371/journal.pone.0046885. Epub 2012 Oct 3.
Armugam, A., Koh, DC., Ching, C.S., Chandrasekaran, K., Kaur, P., Jeyaseelan, K., 2012. Pro-domain in precursor nerve growth factor mediates cell death. Neurochem Int. Jun;60(8):852-63. doi: 10.1016/j.neuint.2012.03.006. Epub 2012 Mar 28.
Besirli, C.G., Wagner, E.F., Johnson, E.M. Jr., 2005. The limited role of NH2-terminal c-Jun
phosphorylation in neuronal apoptosis: identification of the nuclear pore complex as a
potential target of the JNK pathway. J Cell Biol; 170: 401–411.
Biswas, S.C., Shi, Y., Sproul, A., Greene, LA., 2007. Pro-apoptotic Bim induction in response to nerve growth factor deprivation requires simultaneous activation of three different death signaling pathways. J Biol Chem. Oct 5;282(40):29368-74. Epub 2007 Aug 16.
Bredesen, D.E., Mehlen, P., Rabizadeh, S., 2005. Receptors that mediate cellular dependence. Cell Death Differ. Aug;12(8):1031-43.
Chao, M.V., 2003. Neurotrophins and their receptors: a convergence point for many
signalling pathways. Nat. Rev. Neurosci. 4, 299–309.
Clewes, O., Fahey, M.S., Tyler, S.J., Watson, J.J., Seok, H., Catania, C., Cho, K., Dawbarn,
D., Allen, S.J., 2008. Human ProNGF: biological effects and binding profiles at
trkA, P75NTR and sortilin. J. Neurochem. 107 (4), 1124–1135.
Coultas, L., Terzano, S., Thomas, T., Voss, A., Reid, K., Stanley, E.G., 2007. Hrk/DP5 contributes to the apoptosis of select neuronal populations but is dispensable for haematopoietic cell apoptosis. J Cell Sci,120:2044-2052.
Deshmukh, M., Johnson, E.M., Jr. 1998. Evidence of a novel response to cytoplasmic cytochrome c. Neuron 21, 695–705.
Dudek, H., Datta, SR., Franke, T.F., Birnbaum, M.J., Yao, R., 1997. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275: 661–665.
Harris, C.A., Johnson, E.M. Jr., 2001. BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J. Biol. Chem., 276, 37754–37760.
Harrington, A.W., St Hillaire, C., Zweifel, L.S., Glebova, N.O., Philippidou, P., Halegoua, S., Ginty, D.D., 2011. Recruitment of actin modifiers to TrkA endosomes governs retrograde NGF signaling and survival. Cell. Aug 5;146(3):421-34. doi: 10.1016/j.cell.2011.07.008.
Harrington, A.W., Leiner, B., Blechschmitt, C., Arevalo, J.C., Lee, R., Morl, K., Meyer, M., Hempstead, B.L., Yoon, S.O., Giehl, K.M., 2004. Secreted proNGF is a pathophysiological death-inducing ligand after adult CNS injury. Proc. Nat. Acad. Sci. U.S.A. 101, 6226–6230.
Hartikka, J., Hefti, F., 1988. Development of septal cholinergic neurons in culture: plating density and glial cells modulate effects of NGF on survival, fiber growth, and expression of transmitter-specific enzymes J Neurosci. Aug;8(8):2967-85.
.
Hübner, A., Barrett, T., Flavell, R.A., Davis, R.J., 2008 Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell. May 23;30(4):415-25. doi: 10.1016/j.molcel.2008.03.025.
Hughes, R., Kristiansen, M., Lassot, I., Desagher, S., Mantovani, R., Ham, J., 2011. NF-Y is essential for expression of the proapoptotic bim gene in sympathetic neurons. Cell Death Differ. Jun;18(6):937-47. doi: 10.1038/cdd.2010.166. Epub 2010 Dec 17.
Jansen, P., Giehl, K., Nyengaard, J.R., Teng, K., Lioubinski, O., Sjoegaard, S.S., Breiderhoff, T., Gotthardt, M., Lin, F., Eilers, A., Petersen, C.M., Lewin, G.R., Hempstead, B.L., Willnow, T.E., Nykjaer, A., 2007. Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat. Neurosci. 10, 1449–1457.
Kristiansen, M., Menghi, F., Hughes, R., Hubank, M., Ham, J., 2011. Global analysis of gene expression in NGF-deprived sympathetic neurons identifies molecular pathways associated with cell death. BMC Genomics. Nov 8;12:551. doi: 10.1186/1471-2164-12-551.
Lee, R., Kermani, P., Teng, K.K., Hempstead, B.L., 2001. Regulation of cell survival by
secreted proneurotrophins. Science 294, 1945–1948.
Levi-Montalcini, R. 1987. The nerve growth factor 35 years later. Science. Sep 4;237(4819):1154-62.
Ley, R., Ewings, K.E., Hadfield, K., Cook, S.J., 2005. Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell Death Differ;12:1008–1014.
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S.M., Ahmad, M Alnemri, E.S., andWang, X. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489.
Lipscomb, E.A., Sarmiere, P.D., Crowder, R.J., Freeman, R.S., 1999. Expression of the SM-20 gene promotes death in nerve growth factor-dependent sympathetic neurons. J Neurochem. Jul;73(1):429-32.
Masoudi, R., Ioannou, M.S., Coughlin, M.D., Pagadala, P., Neet, K.E., Clewes, O., Allen,
S.J., Dawbarn, D., Fahnestock, M., 2009. Biological activity of nerve growth factor
precursor is dependent upon relative levels of its receptors. J. Biol. Chem. 284
(27), 18424–18433.
Müller, M., Triaca, V., Besusso, D., Costanzi, M., Horn, J.M., Koudelka, J., Geibel, M., Cestari, V., Minichiello, L., 2012 Loss of NGF-TrkA signaling from the CNS is not sufficient to induce cognitive impairments in young adult or intermediate-aged mice. J Neurosci. Oct 24;32(43):14885-98. doi: 10.1523/JNEUROSCI.2849-12.2012.
Nikoletopoulou, V., Lickert, H., Frade, J.M., Rencurel, C., Giallonardo, P., Zhang, L., Bibel, M., Barde, Y.A., 2010. Neurotrophin receptors TrkA and TrkC cause neuronal death whereas TrkB does not. Nature. Sep 2;467(7311):59-63. doi: 10.1038/nature09336.
Nykjaer, A., Lee, R., Teng, K.K., Jansen, P., Madsen, P., Nielsen, M.S., Jacobsen, C., Kliemannel, M., Schwarz, E., Willnow, T.E., Hempstead, B.L., Petersen, C.M.,
2004. Sortilin is essential for proNGF-induced neuronal cell death. Nature 427,
843–848.
Okazawa, H. Estus, S., 2002. The JNK/c-Jun cascade and Alzheimer's disease. Am J Alzheimers Dis Other Demen ; 17:79-88.
Peng. J., Andersen, J.K., 2003. The role of c-Jun N-terminal kinase (JNK) in Parkinson's disease. IUBMB Life; 55:267-271.
Putcha, G.V., Moulder, K.L., Golden, J.P., Bouillet, P., Adams, J.A., Strasser, A., Johnson, E.M., 2001. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron, 29:615-628.
Putcha, G.V., Johnson, Jr. E.M., 2004. Men are but worms: neuronal cell death in C elegans and vertebrates. Cell Death Differ; 11: 38–48.
Sobottka, B., Reinhardt, D., Brockhaus, M., Jacobsen, H., Metzger, F., 2008. ProNGF
inhibits NGF-mediated trkA activation in PC12 cells. J. Neurochem. 107 (5), 1294–1303.
Sobreviela, T., Clary, D.O., Reichardt, L.F., Brandabur, M.M., Kordower, J.H., Mufson, E.J., 1994. TrkA-immunoreactive profiles in the central nervous system: colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J Comp Neurol. Dec 22;350(4):587-611.
Song, W., Volosin, M., Cragnolini, A.B., Hempstead, B.L., Friedman, W.J., 2010. ProNGF induces PTEN via p75NTR to suppress Trk-mediated survival signaling in brain neurons. J Neurosci. Nov 17;30(46):15608-15. doi: 10.1523/JNEUROSCI.2581-10.2010.
Tauszig-Delamasure, S., Yu, L.Y., Cabrera, J.R., Bouzas-Rodriguez, J. Mermet-Bouvier, C. Guix, C., Bordeaux, M.C., Arumäe, U., Mehlen, P., 2007. The TrkC receptor induces apoptosis when the dependence receptor notion meets the neurotrophin paradigm. Proc Natl Acad Sci U S A. Aug 14;104(33):13361-6. Epub 2007 Aug 8.
Towers, E., Gilley, J., Randall, R., Hughes, R., Kristiansen, M., Ham, J., 2009. The proapoptotic dp5 gene is a direct target of the MLK-JNK-c-Jun pathway in sympathetic neurons. Nucleic Acids Res. May;37(9):3044-60. doi: 10.1093/nar/gkp175. Epub 2009 Mar 20.
Whitfield, J., Neame, S.J., Paquet, L., Bernard, O., Ham, J., 2001. Dominant-negative
c-Jun promotes neuronal survival by reducing BIM expression and
inhibiting mitochondrial cytochrome c release. Neuron, 29:629-643.
Wyttenbach, A., Tolkovsky, A.M., 2006. The BH3-only protein Puma is both necessary and sufficient for neuronal apoptosis induced by DNA damage in sympathetic neurons. J Neurochem. Mar;96(5):1213-26.
You, H., Pellegrini, M., Tsuchihara, K., Yamamoto, K., Hacker, G., 2006. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med 203: 1657–1663.


Comments